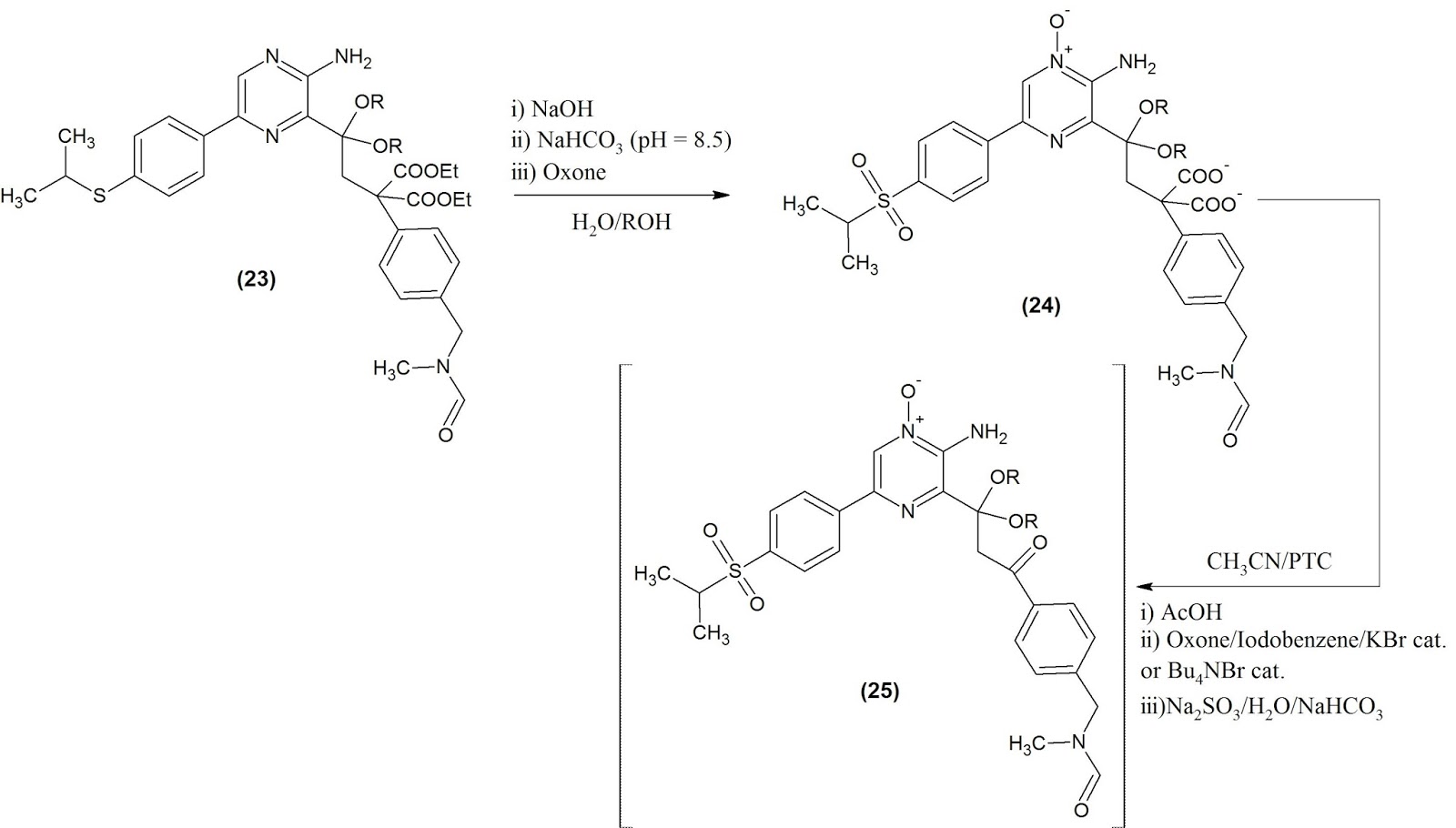
The original route published by Vertex begins with the already synthesised pyrazine core (9 to 12 steps depending if the boronic acid is bought or synthesised) with 2 metal catalyzed cross-coupling. This route is obviously the shorter access to the target, since building the pyrazine core would involve more steps.
I had made an alternative route proposal which avoids cross-coupling reactions in a previous post, it is of course longer than the original route, since this route imply the pyrazine core synthesis by a convergence strategy. Chemically interesting due to some challenges, this route is of course less interesting economically speaking due to the number of steps, but it could give some ideas for others syntheses. The post is available here (Alternate route proposal to VX-970 an ATR Inhibitor (for chemistry fun only) or by scralling down if your are at the index of this blog.
Considering these facts, i have chosen optimizing the route to the chloro oxime synthon only, especially the starting material. There are three methods, one is probably the most feasible, the second must be studied, and the third use solid supported synthesis with the ChemMatrix® resin. The process cost could be potentially 1096$/kg (166$/mol) cheaper on starting material price. But an investment in time is necessary, since a differentiation of reactivity must be done on the starting material.
Complete proposal here
Process optimization proposal

Solid supported version

Original route published by Vertex

Complete proposal here
Disclaimer: This is some personal works on paper only, i have no responsibility in any way if somebody would try this route and has all sort of troubles, including but not limited to: injuries and money loss. This is for experienced chemists only, and tests must be conducted in a suitable lab only.
But if my work is used to synthesize the targeted molecule described here, please, send a word, even if it fails, chemistry is always an experimental science. This will make me pleased, thank you.
© David Le Borgne, 2015, specialist in chemical process development and optimization.